ˎƷ�O(ji��n)���Ǐ��s�ģ��c���ҵİl(f��)չ�v�̡�����������J֪�v�̡��ƌW�ͼ��g�ֶεİl(f��)չ�����P(li��n)��
���鱾ƪՓ�����ģ�
����[ժҪ]2013��2���ӵķ���ˎ�|��һ�����u�r�����LJ���ʳƷˎƷ�O(ji��n)���������֣�CFDA�� ���(zh��)�Ї���ˎƷ��ȫ“ʮ����”Ҏ(gu��)�����_չ��һ���Ҫ�������Ć����������ѽ�ȡ����һЩ�Mչ�����Դ��ں�ҕˎƷ�|���wϵ����Ҫ�ԡ�ȱ�����������Ƅ������w���ܳ�������Ҫ�u�r������3���}�����������ƽ�Q�����o���_��“ʮ����”Ҏ(gu��)�����O����Ŀ�ˣ�Ҳ���o������ˎ������ȫ�[�����o�҇��������������Ϸ�����u����ؓ��Ӱ푣�ʹ�t(y��)���ƶȸĸ�������y����������(j��)�҇����飬�����ˇ��H�Ϸ���ˎ�|���u�r���g���M���ҵ�����������˽�Q2�����}��2헾��w���h�����c�����ġ�ˎƷ����������ӆ������܉��������Ŀǰ������ȱʧ�������P��Ҫ����x�� �煢���Ƅ�������ˎ�� �Լ�����ˎһ�����u�r���P�Iԭ�t��Ҫ��Ҏ(gu��)���ڡ�ˎƷ���������У�������u�r�����ĬF(xi��n)�з��������������(y��u)·���y���䌍���t��CFDA���_���TҎ(gu��)�£��������������Ƅ��wϵ�����_Ŀǰ����ˎһ�����u�r�����ķֲ���ʩ�k�����������M·�������C�u�r�wϵ�ƌW���������ȿ��Ա��C�҇����������ϰ�ȫ��Ч���|���ɿص�ԭ�м�����ˎƷ�����Ƅ��҇���ˎ�a�I(y��)�ڇ��H���l(f��)չ�C�������w��������Kռ�I���H�Ј���
����
����[�P�I�~] ����ˎ�� �|���u�r�� �w���ܳ������� �������
����
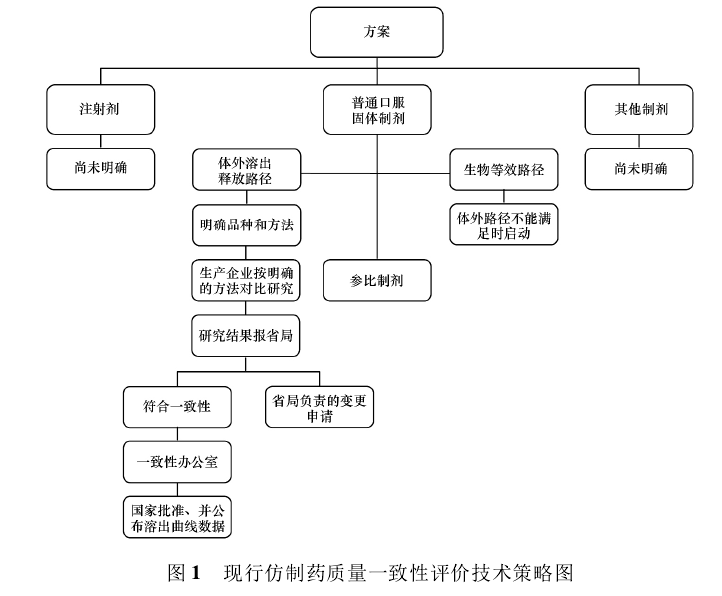
����1�F(xi��n)�з���ˎ�|��һ�����u�r���������ͼ��g����
����
����1. 1����ˎ�|��һ�����u�r�k��������ʽ�l(f��)������Ϣ �҂����Ї�ʳƷˎƷ�z���о�Ժ�Ķ����W(w��ng)վ����ˎ�|��һ�����u�r�k���ҹپW(w��ng)�z����������ʽ���_�l(f��)�����P�ڷ���ˎ�|��һ�����u�r���������P��Ϣ���� CFDA�k���d�P��2013��ȷ���ˎ�|��һ�����u�r�����о��΄յ�֪ͨ�� ʳˎ�O(ji��n)�kˎ����[2013]38̖�� ,��������ˎƷ�z���C����75���ڷ����w�Ƅ��w���ܳ������_չ�о���������һ�����u�r���� ���� �� ˎ �| �� һ �� �� �u �r �� �� �� �������� ���ڷ����w�Ƅ������Ƅ��_��ԭ�t�� �ݰ������͡���ͨ�ڷ����w�Ƅ��ܳ������y���c���^ָ��ԭ�t�� �ݰ��������� 5��ˎƷ�� �}�ᰱ����Ƭ���}���������Ƭ���}�����f��ƽƬ���^������Ƭ����ʯ�������� �� Ƭ ���� �� �� �� һ �� �� �u �r �� �� �� �ݰ���[1].
����
����1. 2�F(xi��n)���γɵļ��g���� �C�ϬF(xi��n)�Ѱl(f��)������Ϣ���R���γ����¼��g���ԈD��Ҋ�D1.
����
����
����
�������g���������ָ������£� �Գ�Ҏ(gu��)�ڷ����w�Ƅ����Ӵ�헹�������ˎƷ���w���ܳ������ı��^�����҇�����ˎ�|��һ�����u�r����Ҫ������ Ҳ��Ŀǰ�����l(f��)����Ψһ������ ,���u�r�ķ���ˎ���c�����Ƅ��M���ܳ������ı��^�����Ѕ����Ƅ�����I(y��)“������Ո-�����u��-��������”�ķ�ʽ�a����ˎƷ�z���C��ؓ؟�w���ܳ��ķ����W�о���
����
����2�F(xi��n)�з����ͼ��g���Եć��H���^����
����
����2. 1����ˎ���҇����ɡ���Ҏ(gu��)��Ҏ(gu��)���ϵĶ��x���a����Ӱ� �҇���ˎ�ИI(y��)�İl(f��)չ�Ǐķ��ƚW�������ѽ����е�ˎƷ�ġ��ܴ˚vʷ���c�Ŀ��^Ӱ푣���ˎ�ͷ���ˎ�ķ��ɶ��x�е��Д�ԭ�t���Пo���҇����к�ˎƷ�˜��Пo����ˎ�䡣���ߵĶ��x1�ٌ�����r������ˎ���R���о���(sh��)��(j��)��֧���@һ��ˎ��ȫ����Ч��ֱ���C��(j��)�������C���m(x��)�ķ���ˎ�c�@һ��ˎ������ͬ����Ч�ԺͰ�ȫ�Ե��P�I��ʹ����ˎ�c�@һ��ˎ���R����(sh��)��(j��)�����B�ӡ����ڌ���������δ�ڿ��]����֮�ȣ��]�н��������B���ƶȣ���ʹ���a���е���ˎ�����Ъ�ռ�ԡ�ͬһ������ֻҪδ���Ї����У��ڷ����϶����Ո�˶��Н��ګ@����ˎ���еę������M��ˎƷ�˜��cˎƷ�|������һ��(li��n)ϵ�������ڃH��ˎƷ�˜��Ƿ�����ˎ���������ˎ���x�еĻ���ԭ�t����δ���]ǰ����2�l���أ���ʹ���еķ���ˎ�H��ˎƷ�˜ʌ����cˎ��˜ʱ�����һ�£����ڷ����ƶȌ���ȱ��������Ҏ(gu��)������ȫ���|�������c��������ȫ�Ժ���Ч�Է����c�ѽ����е���ˎ�� ������Ψһ�ģ�Ҳ�����Ƕ�ң� һ���Լ����`�з���ˎ�c��ˎ���R����(sh��)��(j��)���B�ӡ�
����
����2. 2����ˎһ���ԵăȺ� �ڽ����������ܷ���ˎ������ˎ��ɼ��ԡ�����ˎƷ֧��ؓ���r�������l(w��i)���M����WHO�� ������ˎ�������飺 ����ˎ�ǿ��cԭ��ˎ���Q��ˎƷ��һ����ԣ���ԭ��ˎ�Č����^�ڻ��Ј���ռ�ڽY������ˎ�S�����ڲ���ԭ��ˎ�S�S�ɵėl�������a[2].
����
��������FDA�C���ġ���Ƥ�����LJ��H���J��һ����Ҏ(gu��)�ƌW��Փ[2].�@��1979������r�Ρ��v��34��Č�Փ��Ҫ�Á��C����FDA���ʵġ��cԭ�ЮaƷ����һ���ԵĮaƷĿ䛡��@����Փ����ˎ�W��Ч���ί���Ч�@2����������w�U��һ���ԵăȺ���ˎ�W��Ч��횝M�����l���� ����ˎ�cԭ��ˎ������ͬ�Ļ��Գɷ֣������cԭ��ˎ�ڄ��͡�������Ҏ(gu��)��ʹ��;���ȷ�����ͬ�� �ί���Ч��횝M�����l���� ���ȝM��ˎ�W��Ч�����ί��cԭ��ˎ���f������ͬ���m���Y��Ⱥ�r����һ�ӵ��R����ȫ�Ժ���Ч��[2].FDA�ڡ���Ƥ������Ҏ(gu��)�����ί���ȵķ���ˎҪ�M������5�l���wԭ�t�����cԭ�ЮaƷ����һ�ӵİ�ȫ�Ժ���Ч�ԡ���ˎ�W��Ч���������Ч�� ��ˎƷ���������������Ч�����W�Ć��}�����m�І��}����Ӱ������Ч�ĽY���ж��������m��?sh��)��f�������ݰ���c GMP��Ҫ��M�����a[2].�@5�Ӻ��xĿǰ�LJ��H�Ϲ��J�ķ���ˎһ���ԵĘ������أ�c GMP1PIC/s,���º��Q“���һ����Ҫ��”����Ŀǰ���oՓ��WHO��ˎƷ��ُ��߀�LJ��H�Ͽ���aƷ�J�C���Լ��҇�������ˎ��I(y��)�ڱ����͚W����Ո����ˎ��ע�����У����ǰ����@“���һ����Ҫ��”������(zh��)�еġ�
����
����2. 3����ˎһ���ԵĘ˜�---�����Ƅ� ����ˎˎ�W��Ч���ί���Ч�Ę˜ʣ� �������Ƅ��� �������ׂ����Г��������R����(sh��)��(j��)朵���ˎ�����������@���_��Ψһ�ķ��������Ƅ���reference listed drug���ĭh(hu��n)��(ji��)���Է��ɵ���ʽ����Ҏ(gu��)���������_���롶��Ƥ�������ښW�ˣ��_�����������Ƅ��ĭh(hu��n)��(ji��)Ҳ���ԚW����������ʽ����Ҏ(gu��)�����������Ƶ�ˎƷ����dzɆT�����ѽ�����8�����ϵ�ij�����wˎƷ�����џo�Ј������ں͔�(sh��)��(j��)���o�ڡ��ڰĴ��������@���_�����������Ƅ��ĭh(hu��n)��(ji��)Ҳ������Ҏ(gu��)������ָ�����ˎƷע�Լ���ARTG�����z�����ǣ�Ŀǰ�҇��ķ��ɛ]�����_Ҏ(gu��)������ˎ�ą����Ƅ������]�Ќ���һ���������е���ˎ����Ψһ�����Ƅ����F(xi��n)�з��Ɍ������Ƅ�Ҏ(gu��)���IJ��㌧����Ŀǰ�����е�18�f��ˎƷ������̖�ڷ����ϸ��Ԫ�������̖��ӛ�d�aƷֻ�c��Ո���з����Pϵ����̖���dͬһƷ�N�ĮaƷ֮�g�� ��ͬ����� �������κεķ��ɺͿƌW��(sh��)��(j��)���P(li��n)�ԡ�����ֻҎ(gu��)���@Щ��̖��ӛ�d�aƷ�Ę˜ʱ�횷��χ��Ҙ˜ʡ�ͬ�r�����@Щ�˜��������µķ���ˎҲ���ǺϷ��ġ��@һ���s�ĬF(xi��n)�����o���η���ˎ�|��һ�����u�r����������D�ķ��ɡ��ƌW���Ј��������O(ji��n)�ܲ���������Ӱ푵ȶ�������(zh��n)��
����
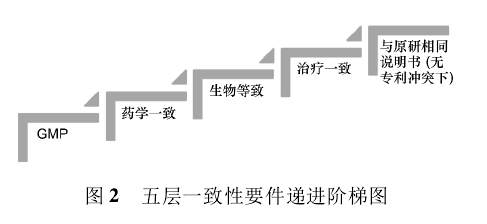
����2. 4��Cһ���Եļ��g·������������Ч�� ǰ�����һ����Ҫ�������D2���f�M�A�݈D���ʬF(xi��n)��
����
����
����
�������ȣ�������GMP�������һϵ���cˎƷ���o�ϡ��Ƅ����Ƃ䡢�ϳɡ��Ŵ�����a����C���|�ؼ����з����Ҏ(gu��)�Ե�һ���ԡ�������Ƅ���̎�������͡�Ҏ(gu��)������ˎ�Wһ�¡��ٺ����������Чȥ��C�����^��C���������Ч�����W�ϟo���g���}���m�І��}����Ӱ푽YՓ���ж����ί���Ч�����ʹ���m��?sh��)��f�����_���cԭ�ЮaƷ���ί��Ͽ����Q����ͬ���Һ͵^(q��)��ˎƷ�O(ji��n)�������m�в�ͬ��������ǰ�����һ����Ҫ�����鼼�g���Եĺ��ġ�
����
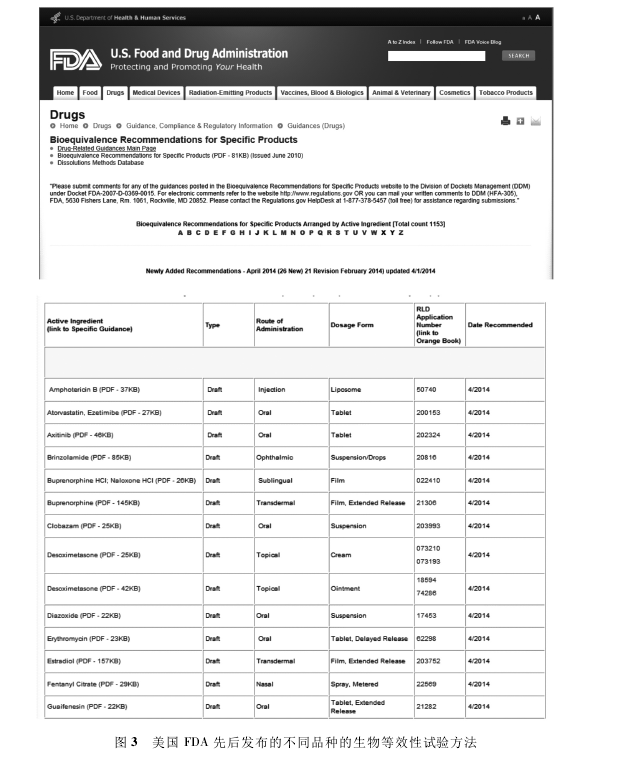
�������g�����е������Ч��ԇ��ǰѷ���ˎ��ˎ�Wһ�£� �|���� ���ί�һ�£� ��Ч�Ͱ�ȫ�� �cԭ�ЮaƷ�����B�ӵ��P�I�� �����Чԇ�ą����Ƅ�����Ƿ���Ҏ(gu��)���ġ����R����ȫ��Ч��(sh��)��(j��)֧�ֵ�ԭ����ˎ�� ,�����Ҳ�Ǻ����еĺ��ġ�����FDA�نT���ӏ���ʿ[3]�ڡ��Ї�̎��ˎ���s־���@���u������������ˎ�Ј�����ķ�Ҏ(gu��)·��ANDA�����ڵ���2�����O���������Ч����ˎƷ��ȫ�Ժ���Ч�Ե�һ�����õ����ָ�ˡ����ڽ�����Ⱥ��ȡ�õ������Ч���о���(sh��)��(j��)�������ǵ�ͬ�ġ��鱣�C��ͬ���������ԡ���ͬ���w���մ��x����ͬ�ί�����ˎƷ���@һ�P�I�B�ӵĿƌW�ɿ�������2014��4������FDA�Ⱥ�l(f��)����1 000������ͬƷ�N�������Ч��ԇ���� Ҋ�D3��[4].
����
����
����
�������H������ˣ�����ˎƷ�O(ji��n)���ƶȰl(f��)�_�ć��Һ͵^(q��)���oһ����ز��������Ч��ԇ������B��ˎ�Wһ�º��ί�һ�µ�“Linker”.����Ŀǰ�����о�δ�l(f��)�F(xi��n)�κ·��һ�^(q��)�ıO(ji��n)�ܙC�����]ʹ���w���ܳ��������^�ķ��������@���P�I��“Lin-ker”.���� �� �J���� �� �� �� �� �� �� �W �� � ϵ �y(t��ng)��biopharmaceutics classification system,BCS�� �� �������ͼ��g���M����Ҳ���l(f��)���g�����ϵ�һЩ�µ�̽�����@Щ̽����Ҫ�۽���ͨ�^�������īI��ԇ��о�ȫ�����������ԭ��ˎ���ܽ��ԡ����w�ȵĝB�ԡ�ˎ�ӌW��ˎ����ί�����ˎ����ί��I���o�ϵ�����õ���Ϣ�������Щˎ������ڼ��g��������������Ч��ԇ�����w���ܳ��������档��WHO��ί�У����Hˎ����(li��n)�ˣ�FIP�� (li��n)�����P�ČW�g�C���ڴ��I�����˴����Ĺ���������W(w��ng)վ���_�l(f��)���ˌ�Փ��monographs��������2014��4�£��Ѱl(f��)����Փ41ƪ����1�г���FIP��WHO�������O(ji��n)�ܙC�����h�Ŀɿ��]��������Ч���о���ͨ�^�w���ܳ�����Cһ���Ե�Ʒ�NĿ�[5].
����
��
![FIP��WHO�������O(ji��n)�ܙC�����h�Ŀɿ��]��������Ч���о���ͨ�^�w���ܳ�����Cһ���Ե�Ʒ�NĿ�](/uploads/allimg/161022/1-161022113525945.png)
����
����ֵ��ע����ǣ��@41��Ʒ�N��FIP��피�W�g�C����ͨ�^������ԇ���īI�����о���֔���_���ģ��������H���鼼�g�����еĘO�ٔ�(sh��)���⣬�������Ч��ԇ���Ȼ�LJ��H���J����C����ˎһ���Ե��ձ�ƌW�ֶ��е��P�I��
����
�������η���ˎ�|��һ�����u�r���g���ԈD�@ʾ�� Ҋ�D1�� ,������ͨ�ڷ����w�Ƅ�����Ҫ���w�⌍�ķ�����ɡ���ĿǰΨһ�l(f��)���ļ��gָ��ԭ�t����ͨ�ڷ����w�Ƅ��ܳ������y���c���^ָ��ԭ�t�� �ݰ������������w�ⷽ����ԓ��ָ�Ƅ����ܳ����������η���ˎ�|��һ�����u�r�ļ��g������������������r����һ��Ҏ(gu��)�t��ֱ�����w���ܳ�����Linker��ȱ���ƌW�C��(j��)֧�ֵġ����ǣ��҂��z���ؿ�����CFDA��ʳˎ�O(ji��n)�kˎ����[2013]38̖�ĵķ�ʽ���ʡˎ�O(ji��n)�ֺ��Ї�ʳƷˎƷ�z���о�Ժ���_���΄�֪ͨ��Ҫ������75��ˎƷ��������w���ܳ������W���о����҂���CFDA��1�ڰl(f��)����Ҫ���ˎ�zԺ�� ���� ���w���ܳ������M���u�r��Ʒ�NĿ��c����FIPĿ��M���ˌ��ȣ��l(f��)�F(xi��n)2��Ŀ��ЃH��14��Ʒ�N�دB���دBƷ�NĿ�Ҋ��2.
����
����

����
����2. 5���H���ȷ��� ͨ�^���Ϸ����������η���ˎ�|��һ�����u�r������ȡ�ļ��g�����c���H��ѭ�ļ��g���Ԍ���Ҋ��3.
����
����

����
����3�F(xi��n)�з����ͼ��g�������R���L�U����
����
�����W�������F(xi��n)��ˎƷ�O(ji��n)���ƶȽ��v�˽�����İl(f��)չ�v�̡�ˎƷ�O(ji��n)���������ƶȺ�����ؙܿC��ʮ�֏��s��ȫ���M��ʹˎƷ���аl(f��)�����a�����γ��ˏ��s��ȫ��机ͱO(ji��n)��朡�ͬ�r����ͬ�ƶ��w���µĻ��ߌ���ˎ��ȫ�ԡ���Ч�ԵĻ����������и߶ȵ�ͬ�|�ԣ� �e�Ƿ���ˎ�� ,�@��ʹ��ȫ��ˎƷ�O(ji��n)�ܣ� �e�Ƿ���ˎ�ıO(ji��n)�ܣ� ������������څͬ�ķ���ʮ�˴���҇������Mһ���Զ��ĸ�ěQ�ĺ����ġ���ǰ���Ї��ѳɞ�ȫ��ڶ����w���������t(y��)ˎ�Ј�����׃ˎƷ��ȫ�΄��ѳɞ�����������Ĺ��R�����ǣ��҂������ɓ�������^�m(x��)���լF(xi��n)�еķ����ͼ��g�������M����ˎ�|��һ�����u�r���������ܕ��oδ���t(y��)ˎ�����I��ĸĸ��c�����������y���o������������Σ�C���п�����K�Ї���ˎ�ИI(y��)�l(f��)չ�͇��H���M�̡�
����
����3. 1����ˎƷ����������ӆ�����ĝ���Ӱ푡�ˎƷ����������ӆ�ѽ��M��{������Ӌ�������ѽ��M�댍ʩ�A�Ρ���ǰ����������������ķ���ˎָ�����ԭ����ˎָ������ɷ���ˎ���߂��ί���Ч�Ϳ�����Եĸ������ص�Q�@һ���}��Ҫͨ�^���ɵ���ӆ�����ơ���ˣ����������Ű��Sδ���Ј���������ƺ��t(y��)ˎ�����İl(f��)չ���������Q�ɆT�����҇��ڱ��Ρ�ˎƷ������������ӆ�����Е���˼���ͽ�������(chu��ng)���c�l(f��)չ�ķ����ƶȣ����ɬF(xi��n)�е����Ƿ��Ї��Ҙ˜ʄ�����ˎ�ͷ���ˎ���D׃?y��u)��c���H��܉��֪�R�a����R����ȫ��Ч��(sh��)��(j��)�턝����ˎ�ͷ���ˎ�������η���ˎ�|��һ�����u�r��������I(y��)���������������M��/�ҽM�u�h�@�N�����О�ķ����_�������Ƅ��Խ�Q�vʷ�z���}���������o�F(xi��n)���Ј������δ�������ĸ���ϵK���ھ��w���u�r�����W�ϣ����w���ܳ����LJ��H���R�������Ч���_չ��Ҳ����ɲ��ݵ��ć��Hؓ��Ӱ푡�
����
����3. 2���f(xi��)�{�F(xi��n)�в��T�g���ߺ����P�ĸ��ʩ�������y �t(y��)���ƶȡ�ˎƷ�����ƶȺ��t(y��)���Ǹ����������J�������s������������������ա�Ŀǰ�漰�t(y��)�ĵ������A���͇����Ұl(f��)չ�ĸ�ί�T�������A���͇������l(w��i)����Ӌ������ί�T�������A���͇������YԴ��������ϲ���CFDA��ʮ�ׂ����T������ί����(j��)���ܷ��^����ˎƷ�r�����t(y��)�����UĿ䛡�����ˎ��Ŀ䛡�ˎƷ�И�Ͷ�˵��I���������������ա���Ŀǰ����ˎ�|��һ�����u�r�����������������κ��c�@Щ���T�Ĺ���㕽ӡ����^�ϣ�CFDA���_�J�ą����Ƅ�������ˎ���|��һ�µķ���ˎ���|����һ�µķ���ˎ���κ�׃��������Ӱ푵�������ί�Ĺ����������ƶ������ƶ������ߡ���ȱ��ǰ�����ɻ��A�����H���g���Ի��A�������ܲ��T(li��n)�ӵĻ��A�����M������CFDA���������T�����L�U��
����
����3. 3�c���H���M�O(ji��n)�܌��`�IJ ����Ժ�C��“ˎƷ��ȫҎ(gu��)��”�����_Ŀ����“ʮ����”ĩˎƷ��ȫ�c���H��܉�����䌍��Ҏ(gu��)���ĬF(xi��n)�з���ˎ�|��һ���Թ��������ͼ��g����߀ȱ���c��ǰ���H��܉�ļ��g�wϵ֧�֣� ��ǰ����Ҏ(gu��)��������ɶ��x����IJ�Լ����g·���IJ�⣬�ڿƌW���棨 ���gָ���wϵ�� �^�m(x��)���լF(xi��n)�з����ͼ��g�������M����Ҳ�������L�U�����y�����@����ˎ�ć��Hע�Ի��|���ܿأ�Ŀǰ���H���J�ļ��g�wϵ��2����һ��ICH,����WHO���A�J�C�wϵ��ICH��20���o90������ɚW��������3���͵^(q��)����ˎ�I(y��)�f(xi��)���ͱO(ji��n)�ܲ��T��ͬ�M�ɵć��H����ˎ��f(xi��)�{�C�ơ��@һ�f(xi��)�{�C�ƹ�ͬ�ƶ���ˎƷ�Ј���������ѭ�ĿƌW�˜ʣ��քe��ICH�C����Q/S /E /M 4��ϵ�е�ָ��ԭ�t�鼼�g�˜ʡ��҇�Ŀǰ߀δȫ����bICH�ƶ������PNDA��ע�Լ��gҪ���⣬�҇���ǰ�ķ���ˎע��·�����|�������wϵ�cWHO��ˎƷȫ���ُ�A�J�Cϵ�y(t��ng)���Ҳ�����@��ࡣWHO�ȇ��H�M�����҇��M�е�ˎƷȫ���ُ߀̎�ڄ������A�Σ� ������A�J�C�ѽ�ȡ�ó����Y������������M���о���Q�@һ���}�����ǰ��լF(xi��n)�еķ����^�m(x��)���M���@�N�����x��Խ��Խ��ͬ�rҲ����WHO�A�J�C���҇����Mһ�����M�a��Ӱ푡�
����
����3. 4���߿��ܳГ����L�U �������ˎһ�����u�r�����o����Ч�غY�����ȫ��Ч�Բ��_�˵ķ���ˎ���������˳��Ј�����ô�҇����ߌ��Г�����L�U�����ȣ����߷����˯�Ч���_�������]�Я�Ч�ķ���ˎ֮���o�����AӋ�į��̃ȵõ����͡��@�����L���ߵ��ί��ڣ����M���߸����ؔ���c���������⣬���ï�Ч���_�˵ķ���ˎ����ʹ�����e�^�S���ί��ڣ���KӰ푻��ߵĿ���Ч���������������ӽo���߽������Σ�������ߣ��Ļ��ߵĽ���Ч�ʽǶȁ���������ȱ�����I(y��)֪�R����ͨ���߶��ԣ����y�ı������Q������ͬ��Ч�ĮaƷ���x������������ί��Լ�������ˎƷ�� �t(y��)��Ҳͬ�����R�@һ�x��ɱ����������|�����_�˵ķ���ˎ���������������a��ˇ���ʹ���˵ͳɱ���ԭ���ϣ��@�ø�����Ј�������(y��u)�ݣ��Ķ����F(xi��n)“�ӎ��������”Ч�������L�ځ����������҇����߫@����Ч�ί��a������Ӱ푡�
����
����3. 5��ˎ�ИI(y��)�������R���L�U ����ˎһ�����u�r�����ĽY��Ҳ�����Ӱ��҇��t(y��)ˎ�ИI(y��)��δ���l(f��)չ����ǰ�������t(y��)���c���ߏ��L�ڵ���ˎ����Е���u�R�e����ȫ��Ч�Բ��_�˵ķ���ˎ������ˌ����aˎƷʧȥ���ģ��Ķ�����ˎ�ИI(y��)ʧȥ���ġ�һ���l(f��)�����@һ�Y����Ҫ��u�֏������ģ���Ҫ������I(y��)���M����ĕr�g�����X�c���������ć��a����Ʒ�ИI(y��)����һ���ܺõ����C����Σ���ǰ�������ڬF(xi��n)�еĶ��r�c�И��w���£���ȫ��Ч�Բ��_�˵ķ���ˎ�Ĵ��ڕ���u���|����(y��u)����ͬ�ˎƷ�����Ј��������ǵͳɱ��c�r����Ǹ������������@�c�҇��������緶�����t(y��)ˎ�ИI(y��)�����Ĵ����аl(f��)�c��߮aƷ�|���DZ������Y�ġ����ߣ��҇��t(y��)ˎ�ИI(y��)�l(f��)չ�ĽK�OĿ����“Made in China,For China;Made inChina,For World”.���@һĿ�˵Č��F(xi��n)��������Ҫ����҇���ˎ��I(y��)���аl(f��)������������Ҫ�������҇�����ˎ�����a�|�������_�����H���Mˮƽ���Ķ��܉������������������һ����c�t(y��)���J�ɺͽӼ{������“ˎƷ��ȫҎ(gu��)��”����������Ŀ�ˣ���“ȫ�����WˎƷ��������Ʒ�˜��_����ӽ����H�˜ʣ���ˎ�˜��������H�˜��ƶ�”.
����
����4ᘌ�����ˎ�|��һ�����u�r�Ŀ���ИI(y��)���h
����
���������ό��҇�����ˎһ�����u�r�����ķ������Կ������҇��������F(xi��n)�з����c���g�����M�м��r���{���c���M���Ķ�����ƌW��Ч���_չ�u�r���������F(xi��n)�з����c���g���Ե��{���c���M���҂����h���ԇLԇ�����(y��u)���M·����������ȡ�(y��u)���M·�����������ɿ˷����������ɇLԇ��(y��u)���M·����
����
����4. 1�(y��u)���M·��---�ġ�ˎƷ���������еķ���ˎ��Ҏ(gu��)�� ���h�c��ˎƷ������������ӆ������܉������Ŀǰ������ȱʧ�������ƶ���헹��������w�Č�ʩ����������ǰ�����������ڬF(xi��n)�з����ƶȌ�����ˎҎ(gu��)���IJ����ƣ�ʹ��һ�����u�r�����Č�ʩȱ�پ��w�ķ�������(j��)��������ڷ���Ҏ(gu��)���ƶ�һ�����u�r�ľ��wҪ�t���ܕ��c�պ�ķ�����ӆ���·����_�γɛ_ͻ����ˣ��҂����h���ɽ���Ŀǰ����������ӆ��ˎƷ���������ĕr�C��������ˎ����Ҫ����x�� �����ˎ��������ˎ�ȣ� �cһ�����u�r��ԭ�t�c���wҪ��Ҏ(gu��)���ڡ�ˎƷ���������@һˎ�·������Ч���ķ����С����������(j��)�@ЩҎ(gu��)���ƶ�һ�����u�r���������w��ʩ�������������������Ƅ��wϵ���@�ӼȞ�˹����춨�˷��ɻ��A������“�����·�”�ķ�����ߣ�ͬ�rҲ�����˹��������c�պ�����_ͻ�Č��ξ��档���⣬�҂��J�飬�M�ܡ�ˎƷ������������ӆ������ʹһ�����u�r�����Ӻ��_չ�����@����Ӱ�“ˎƷ��ȫҎ(gu��)��”�Č�ʩ�����һ�����u�r�����H�H��Ҏ(gu��)���е�һ���֡�һ����δ�_�ɲ���Ӱ����wҎ(gu��)���Č�ʩЧ����
����
����4. 2��(y��u)���M·��---�IJ��TҎ(gu��)���еķ���ˎҎ(gu��)�� ���y���c��ˎƷ������������ӆ������܉���������PҎ(gu��)����ʩ���T���_���TҎ(gu��)�£����_�ֲ���ʩ���^�̡����]����ˎƷ������������ӆ�����漰�������T�c�I��ąf(xi��)�{�������һ�N·���в�ͨ�����ԇLԇ�����PҎ(gu��)����ʩ���T�� ��Ҫ��CFDA�� ���_���TҎ(gu��)��һ�����u�r������ȫ��ϵ�y(t��ng)�IJ��TҎ(gu��)�£�Ҏ(gu��)��һ�����u�r�Ļ���ԭ���c���wҪ���ڱ����ǰ�Č��W�ˡ������c�ձ��ɹ����Ľ�B�����҇��F(xi��n)����J�R�����h���ҿ��Կ��]��һ�����u�r�����ֲ��M�С��༴�����TҎ(gu��)�¿���Ҏ(gu��)�������M����GMPܛ��ϵ�y(t��ng)�������|���wϵһ�����u�r���Ķ���̭һ����ܛ��ϵ�y(t��ng)���ϸ�ķ���ˎ�aƷ�� ����M�����w���ܳ�������ˎ�Wһ�����u�r������̭һ��ˎ�W���_�˵ķ���ˎ�aƷ�� ����M������һ�����u�r����K��̭һ������ˎ�aƷ����݆һ�����u�r���_�˵ĮaƷ�ű��J�����c������ˎһ�¡��@һ·����һ��(y��u)1�ͣ�����ǰһ݆��̭�ĮaƷ�������M��ڶ�݆�u�r���������؏��u�r��
����
����4. 3���҇���ˎ�a�I(y��)���w�������ش����x �҇���ˎ�a�I(y��)Ŀǰ���R����Ҫƿ�i�������аl(f��)����������ȱ��ԭ��ˎ�aƷ���@һƿ�i��ͻ����Ҫ�L�ڡ����m(x��)���������аl(f��)���M�c�˲�Ͷ�룬��һ��һϦ�����_�ɡ����҇���ˎ�a�I(y��)���ж��ڿ��������Ć��}�ǣ��҇�����ˎ�aƷ�|������R���a���^ʣ�������^�ȶ������ʵ��¡����^�ȸ����D�����������g�Լ��^�͵ıO(ji��n)��Ҫ����m�ϱ�����С�ͷ���ˎ��I(y��)�Ե̓r�_���Ј��IJ��ԣ��������ڰl(f��)չ���|������ˎ�aƷ�ĬF(xi��n)������I(y��)�ڵͼ��g�����ăr���(zh��n)��̎���ӄݣ���K���С��I(y��)������I(y��)���Ј�����
����
��������(j��)�Y���@ʾ��Ŀǰȫ�����ˎ�Ј���Ҏ(gu��)ģ�_800�|��Ԫ������8%���ٶ����L�����Ї�����ˎ�ИI(y��)���L�ٶȞ�25% .��2012��1��1������2016��12��31�յ�5���g��ȫ���ж��_631������ˎ���ڡ��c��ͬ�r����������@ʾ���Ї��t(y��)ˎ�Ј��l(f��)չ?ji��)����?015���Ї����ɞ�ȫ��ڶ����t(y��)ˎ�Ј�����[6].�@Щ��(sh��)��(j��)�Aʾ���Ї�����ˎ�a�I(y��)��ӭ��һ��ǰ��δ�еİl(f��)չ�C�����҇������c��I(y��)�������@һ�vʷ�C�����ƌW��Ч���_չһ�����u�r�������Ķ����M�a�I(y��)������
����
�������F(xi��n)�з����c���g���Ե�ǰ���{���c���M�������҇��ƌW��Ч���_չһ�����u�r������ͨ�^��ʩ���Hͨ�еļ��g���ԣ����F(xi��n)�҇�����ˎ�|���c���H�˜ʽ�܉���@һ�����{�����B�iЧ�����Ǿ�ġ��ć��ȁ������@���ƌW��������̭һ���|�����¡��؏��ʸߵķ���ˎ������߷���ˎ�Ј����жȣ��֏ͷ���ˎ�aƷ�ĺ����������g�������ڰl(f��)չ���|������ˎ�aƷ����I(y��)�������Ը�����ȡ�٣��Ķ������������Mһ�������Լ����аl(f��)������ͬ�r�����|������ˎ�aƷ���{��r����(y��u)�|���Zԭ��ˎ���Ј����~�������ڸ���߫@����Ч�ί����p���t(y��)ˎ�_�N�����õع�(ji��)ʡ�t(y��)���M��֧�����ć��H������ͨ�^��ʩ���Hͨ�е�ע�Լ��gҪ���c�|�����Ƙ˜ʣ�������ȵ؎����҇�����ˎ��I(y��)�߳����T���~����H�Ј����A��ˎ�I(y��)������ˎ�I(y��)�����ɇ����I�ȵ���ˎ��I(y��)�ѽ�ͨ�^�̘I(y��)���`��������ˎ��I(y��)�C�����@һ·���Ŀ������c��Ҫ�ԡ�����ͨ�^���������aƷ�|���_�����H���Mˮƽ�����@������FDA�c�������ұO(ji��n)�ܲ��T���J�C�c���ģ��ѽ�����ԭ��ˎ���ڞ����ɹ��D�͞��Է���ˎ�Ƅ����ڞ������ărֵ朵Ͷ���u��rֵ朸߶��������@Ҳ�������҇���һ������ˎ���������D�͞����ˎ������
����
����5�Y�Z
����
�����vʷ�l(f��)չ�M���л����J֪�ľ����Լ����ڿ��^�M�����z���}�Ľ�Q������һ�����͡�FDA����2014���C���ĵ�34�桶��Ƥ��������Ȼ���_�U���������J���Ŀ����鱻���Ƶ�����(j��)��ˎƷ��������ˎ�� ����(j��)1938��ǰ��Ҏ(gu��)���е�ˎƷ�� �̓H�C����ȫ�Ե�ˎ� ����(j��)1962�귨Ҏ(gu��)���е�ˎƷ�� ,��ֻ��1962�������а�ȫ����Ч���R���C��(j��)��ˎƷ�ſ����鱻���Ƶ�����(j��)��ˎƷ�O(ji��n)���Ǐ��s�ģ��c���ҵİl(f��)չ�v�̡�����������J֪�v�̡��ƌW�ͼ��g�ֶεİl(f��)չ�����P(li��n)���������ڻ��ڱ������������M�������m�������Ľ��}��������������ӆ�����˕r�����ޣ��ƌW���������뱣�C�˷������_���҂����ţ��{��ĸ�ěQ�ĺͿƌW�đB(t��i)�Ȳ�����������R���γɵķ������Ǵ��M�҇���ˎ�ИI(y��)������Ч�l(f��)չ�����F(xi��n)������ˎ��ȫ�ı���֮·��
����
�������n�}��2014�����ʽ��헣�2014��8�¹�ͬ��ɴ��n�}��棬�Ⱥ�õ����P�I�����ИI(y��)���ҵĎ����Ą�e��CFDA���Ї�ʳƷˎƷ�z���о�Ժ���Ϻ���ˎƷ�z��������P�I���͌��Ҍ����n�}�������ˌ��F��ָ����Ҋ���������n�}���ָ���˷����҂���ϲ�ؿ�����2015��11��18�գ�CFDA�l(f��)�����P�������P���_չ����ˎ�|���ͯ�Чһ�����u�r����Ҋ�� ������Ҋ�壩����Ҋ�Ĺ��棨2015���231̖�� ,���“ԭ�t����I(y��)�������w�������Ч��ԇ�ķ����M���u�r�����S��I(y��)��ȡ�w���ܳ���ԇ�ķ����M���u�r�������w���ܳ���ԇ���M���u�r��Ʒ�N���Ժ�߀������ȡ�w�������Ч��ԇ�ķ����M�к��m(x��)�u�r”.����ˎƷ�O(ji��n)�ܲ��T���e�O���Շ��H���ԳֿƌW�l(f��)չ�^���Ќ������÷���ˎ�|��һ�����u�r�������҇�Ҳ���ķ���ˎ������������ˎ�������S���҇�ˎƷ�O(ji��n)���ƶȵ�һϵ�иĸ���Һ���ˎ�a�I(y��)����������ҕ��(chu��ng)�º��|�����҇���ˎ�a�I(y��)�����Rȫ�µİl(f��)չ�A�Σ��҂��������Ų��h�Č����҇�һ���܉�ɞ���ˎ������
����
����[ �� �� �� �I ]
����
����[1] �Ї�ʳƷˎƷ�z���о�Ժ����ˎ�|��һ�����u�r�k���ҡ��P�������^������Ƭ�����f��ƽƬ���}���������Ƭ���}�ᰱ����Ƭ����ʯ�������堖Ƭ��5�NˎƷ�ܳ�����һ�����u�r������ �ݰ��� ��Ҋ��֪ͨ[EB /OL].[2014 - 02 - 21]. http:/ /www. nifdc. org. cn / fzy / CL0622 / .
����[2]Food and Drug Administration Center for Drug Evaluation and Re-search Office of Medical Products and Tobacco Office of GenericDrugs,Approved drug products with therapeutic equivalence eval-uation[EB / OL].[2014 - 05 - 11]. http:/ / www. fda. gov / down-loads / Drugs / Development Approval Process / UCM071436. pdf.
����[3] ���ӏ�����������ˎ�Ěvʷ��׃[J].�Ї�̎��ˎ��2008,7��9�� :47 - 49.
����[4]FDA. Drug Price Competition and Patent Term Restoration Act of1984[EB / OL].[2014 - 10 - 10]. http:/ / www. fda. gov / news-events / testimony / ucm115033. htm.
����[5]International Pharmaceutical Federation��FIP�� ,Biowaiver Monogrphs[EB/OL].[2014 - 05 - 11]. http:/ /www. fip. org/bcs_mono-graphs.
����[6] �w�á��Ї�����ˎ�a�I(y��)ӭ���� ������Ј����_5000�|[EB /OL].[2014 - 07 - 04]. http:/ / money. 163. com / special /view415 / .